La “approvazione del vaccino mRNA” è stata una farsa. Le morti e le lesioni sono reali e intenzionali.
Nota: il video di presentazione di questo materiale è qui. Lo pubblico come articolo scritto poiché molti me lo hanno chiesto.
La totalità delle prove finora raccolte indica che le iniezioni anti Covid-19, così come tutte le altre “contromisure di Covid“, provocano danni intenzionali. Non dedicherò molto tempo alla dimostrazione di questo fatto in questo articolo, ma collegherò ampie fonti di prove documentate. Le iniezioni e le altre “contromisure”, come le chiusure [lockdown], la soppressione di trattamenti precoci efficaci, l’uso eccessivo di oppioidi dietro le porte chiuse degli ospedali e delle case di cura, l’uso eccessivo di remdesivir inefficaci e dannosi (ma molto redditizi), l’uccisione dei pazienti in un ventilatore o semplicemente la loro morte per fame rinchiusi nella “unità covid” dell’ospedale, sono tutte insensate nel contesto della salute pubblica e sono tossiche per progettazione.
I meccanismi di danno sono progettati nelle iniezioni di C-19.
In parole povere, semplicemente addestra le cellule ad attaccare e distruggersi. È solo una questione di esposizione, ovvero di quante istruzioni vengono impartite, dove e in quali quantità nel vostro corpo, a determinare se morirete di infarto o ictus nel giro di pochi giorni o mesi o se vi deteriorerete in modi più lenti e dolorosi (ma redditizi!) a causa di cancro, neurodegenerazione, condizioni autoimmuni o altre forme di morte cronica.
Non c’è sicurezza né efficacia in questi prodotti, anzi osserviamo un numero spaventoso di morti e feriti (VAERS, vSAFE, Eudravigilance, Yellow Card, ecc. contengono milioni di segnalazioni). È stata documentata anche l’efficacia negativa, cioè la propensione a causare malattie da COVID.
Poi c’è una produzione MOLTO scadente: Produzione altamente variabile, non conforme alle cGMP, accompagnata dall’assenza di applicazione delle cGMP da parte di qualsiasi agenzia. Nelle prossime settimane ripubblicherò su questo sottopancia la mia ampia ricerca sull’argomento.
Infine, tutto questo è mantenuto da una politica profondamente maligna in tutto il mondo: bugie del governo, insabbiamento, l’abuso e manipolazione psicologica [gaslighting] dei feriti, persecuzione del dissenso e degli informatori, collusione con i media e finanziamento massiccio e perverso di quanto sopra.
Se non vi rendete conto di tutto questo, Dio vi aiuti, e lo dico con compassione.
Se invece lo vedete, probabilmente a questo punto starete gridando “Lo so!!!”. Perché non è stato fatto nulla al riguardo? Perché tutti i regolatori si comportano così?“. Ecco perché.
Fatti legali fondamentali
(leggete le notizie su Bailiwick di Katherine Watt se volete sopravvivere a questo periodo storico)…
Gli emendamenti del Congresso al FD&C Act del 1938 e al PHS Act del 1944, nel corso dei decenni, hanno eliminato gli standard normativi federali per la produzione e l’uso di prodotti designati dalla FDA per un “uso di emergenza” durante un'”emergenza sanitaria pubblica” dichiarata e mantenuta dall’HHS.
In particolare, 21 USC 360bbb-3(c) “Criteri per il rilascio dell’autorizzazione“: la legge prevede che il Segretario dell’HHS possa rilasciare autorizzazioni per l’uso di emergenza se conclude che:
sulla base della totalità delle prove scientifiche a disposizione del Segretario, compresi i dati provenienti da studi clinici adeguati e ben controllati, se disponibili, è ragionevole ritenere che…
- il prodotto possa essere efficace nel diagnosticare, trattare o prevenire
- tale malattia o condizione; o
- una malattia o condizione grave o pericolosa per la vita causata da un prodotto autorizzato ai sensi della presente sezione, approvato o autorizzato ai sensi del presente capitolo, o concesso in licenza ai sensi della sezione 351 del Public Health Service Act [42 U.S.C. 262], per la diagnosi, il trattamento o la prevenzione di tale malattia o condizione causata da tale agente; e
- i benefici noti e potenziali del prodotto, se utilizzato per diagnosticare, prevenire o trattare tale malattia o condizione, superano i rischi noti e potenziali del prodotto, tenendo conto della minaccia materiale rappresentata dall’agente o dagli agenti identificati in una dichiarazione ai sensi della sottosezione (b)(1)(D), se applicabile;
Si noti che l’autorità che rilascia le autorizzazioni all’uso in emergenza è il Segretario dell’HHS stesso. Non vi è inoltre alcuno standard normativo applicabile per il rilascio, se non l’esclusiva discrezionalità del Segretario HHS nel determinare la “possibile” efficacia. Non è necessario che siano disponibili prove scientifiche a sostegno di questa decisione e non ci sono standard scientifici rigidi per valutare i rischi o i benefici, il “potenziale” è uno standard sufficiente in questo caso. Non ci sono nemmeno criteri di arresto o processi che aggiornino/modifichino la decisione del Segretario dell’HHS se e quando saranno disponibili nuovi dati scientifici. Cosa succede se si rendono disponibili dati scientifici più rigorosi, ad esempio serie di dati più ampie, raccolte per un periodo più lungo, e i risultati contraddicono la decisione presa dal Segretario HHS sulla base di dati piccoli e raccolti frettolosamente? Non esiste un processo legale definito per rivedere tali decisioni.
Di particolare interesse è una categoria speciale di prodotti designati dal Dipartimento della Difesa (DoD) come “prototipi di contromisure“.
– 10 USC 4022(a)(1) – “[Il] Direttore di [] (DARPA), il Segretario di un dipartimento militare, o qualsiasi altro funzionario designato dal Segretario della Difesa può, sotto l’autorità della sezione 4021 di questo titolo, realizzare progetti di prototipi che sono direttamente rilevanti per migliorare l’efficacia della missione del personale militare e delle piattaforme di supporto, dei sistemi, dei componenti o dei materiali proposti per essere acquisiti o sviluppati dal Dipartimento della Difesa, o per migliorare le piattaforme, i sistemi, i componenti o i materiali in uso dalle forze armate“.
Il linguaggio sopra riportato è composto da molte parole che non descrivono nulla di specifico. Si tratta semplicemente di “cose di cui il Dipartimento della Difesa ha bisogno“. Non è necessario sapere cosa siano. Perché sapete già cosa sono: sono armi. Queste sono le cose di cui il Dipartimento della Difesa ha bisogno la maggior parte delle volte.
Ultimo pezzo del puzzle legale:
21 USC 360bbb-3(k): l’uso di prodotti di contromisure mediche (MCM) coperti da EUA, una volta designati come tali dal Segretario della Salute e dei Servizi Umani (10 marzo 2020, retroattivo al 4 febbraio 2020) “non sarà considerato come un’indagine clinica“. 21 USC 360bbb-3(k). Legge EUA, adottata nel 1997 e modificata nel 2003, 2004, 2005, 2013, 2017.
Le contromisure NON sono legalmente prodotti farmaceutici.
La FDA non ha alcuna autorità su di loro e non può applicare alcun regolamento. Fa solo finta di farlo. Questo risponde alla domanda sul perché le autorità di regolamentazione agiscono in questo modo. Stanno recitando. Come in “teatro“.
Il Dipartimento della Difesa è responsabile della produzione di contromisure per il Covid-19:
L’operazione Warp Speed è stata pubblicizzata come uno sforzo “collaborativo” del Dipartimento della Difesa e dell’HHS per produrre vaccini Covid-19 “sicuri ed efficaci“. Tuttavia, secondo l’organigramma, il Dipartimento della Difesa era formalmente il responsabile operativo, mentre l’HHS aveva la posizione di consulente scientifico capo. L’organigramma sottostante è tratto dal documento “VRBPAC-10.22.20-Meeting-Presentation-COVID19-Vaccine-Development-Portfolio“.
In particolare, il livello più alto dell’organizzazione è interamente governativo e comprende tutti i ruoli di supervisione per la produzione, gli studi clinici, la distribuzione, gli affari pubblici, i contratti, la copertura legale (ovviamente!) Ricordate che gli avvocati del DoJ hanno discusso per conto di Pfizer in tribunale, difendendo i loro “segreti commerciali” mentre cercavano di nascondere i dati degli studi clinici per 75 anni. Ora tutto ha senso. Si tratta di un’impresa del governo degli Stati Uniti, in particolare del governo militare degli Stati Uniti. Le farmaceutiche sono un terzo livello in basso in questa organizzazione e “eseguono gli ordini“. Per “dimostrazioni” e “prototipi”. Si noti che “una struttura organizzativa simile supporta lo sviluppo terapeutico“: tutte le contromisure di Covid sono state ordinate e organizzate in questo modo. È tutto gestito sotto la stessa operazione guidata dal Dipartimento della Difesa.
Qui c’è un’ottima ricerca di Whitney Webb (che l’ha vista molto prima di me) che parla della leadership del Dipartimento della Difesa e della proprietà di Warp Speed. Un altro rapporto di STAT ha evidenziato che su circa 90 posizioni di leadership nell’organigramma, solo 29 non erano dipendenti del Dipartimento della Difesa.
Revisione dei contratti del Dipartimento della Difesa per le contromisure Covid:
L’Other Transaction Authority (OTA) è un metodo di contrattazione particolarmente favorito dal Dipartimento della Difesa, che consente di ordinare prodotti altrimenti regolamentati aggirando tali regolamenti, nonché la responsabilità dei contratti governativi standard e altre leggi che regolano la divulgazione e la proprietà intellettuale derivata dalla ricerca finanziata con fondi pubblici. Questo non dovrebbe sorprendere nessuno. “Altro” è una categoria generica che non è un contratto, non è una sovvenzione di ricerca, non è un appalto, eccetera, non è un contratto governativo normalmente regolamentato/responsabile.
Il Dipartimento della Difesa utilizza l’OTA per ordinare “prototipi” e “dimostrazioni” vagamente definiti che non sono soggetti ad alcun controllo normativo. DARPA (all’interno del DOD) e BARDA (tecnicamente all’interno dell’HHS, ma al di fuori della FDA) distribuiscono mega-dollari in varie forme, tra cui finanziamenti di rischio. Il BARDA ha recentemente riferito che nel periodo 2020-2021 ha distribuito 47,5 miliardi di dollari in finanziamenti di R&S per le “contromisure covid“, di cui 33 miliardi di dollari per le iniezioni di covid-19. Molti contratti DOD/BARDA sono stati resi noti in forma redatta. Per avere un’idea, l’intera spesa per la R&S farmaceutica degli Stati Uniti ammonta a circa 100 miliardi di dollari all’anno, quindi la BARDA controlla l’intero settore della R&S farmaceutica (il 50% di un singolo acquirente è più che sufficiente per controllare l’intero settore).
Inoltre, nel rapporto della stessa BARDA, tutte queste ingenti spese erano destinate a “dimostrazioni” o, nel migliore dei casi, a “produzioni su larga scala“, non a “prodotti sicuri, efficaci e conformi alle cGMP” – si veda il linguaggio sul lato destro di questo grafico. Questi tecnicismi e queste curiose formulazioni sono molto importanti.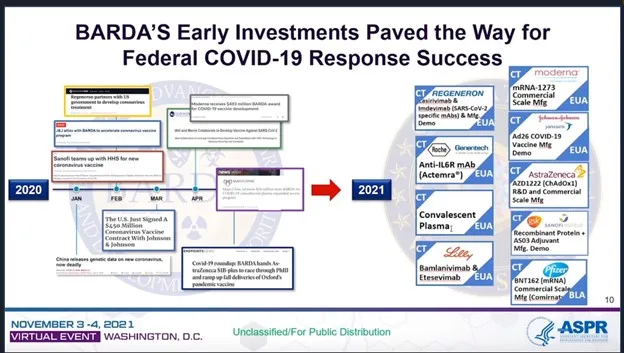
È inoltre importante notare che la BARDA non è un ente regolatore del settore farmaceutico negli Stati Uniti, mentre lo è la FDA. Sono rimasto sorpresa quando ho assistito a un evento pubblico della BARDA (BARDA Industry Day, 15-16 novembre 2022), in cui un rappresentante della BARDA ha affermato che il suo dipartimento RQA (regulatory and quality assurance) ha testato e rilasciato 600 milioni di dosi di [c.d.] vaccino covid e 23 milioni di dosi terapeutiche negli Stati Uniti, oltre ad aver aumentato la “sorveglianza regolamentare dell’industria” e ad aver eseguito controlli di qualità sui produttori. Eh? Vorrei sapere quale legge del Congresso ha trasferito l’autorità della FDA alla BARDA e quando è successo? L’oratore che ha presentato questo materiale è Tremel Faison, direttore della Divisione Affari Regolatori e di Qualità del BARDA.
Si noti che l’url del sito web di BARDA è https://medicalcountermeasures.gov/barda/ . I contratti DOD/BARDA per le “contromisure” sono gestiti da un “manager”. Questo gestore è Advanced Technology International (ATI) – ati.org. Secondo il suo sito web, l’ATI è una “società senza scopo di lucro” (questa sì che è divertente!). Immaginate di “gestire” miliardi di dollari per costruire portaerei, caccia e altri giocattoli del Dipartimento della Difesa! Lasciate che vi spieghi il linguaggio governativo: “Non profit” = “cost plus” = “profitto % garantito x gazillion bucks” = meglio di quanto i peones del settore privato possano mai sognare. Comunque, l’ATI gestisce principalmente consorzi di ricerca e sviluppo per il Dipartimento della Difesa per cose come la produzione di armi, la fusione e la forgiatura dei metalli, la produzione di navi e la tecnologia finalizzata a “contrastare le armi di distruzione di massa (WMD)“. Due di questi consorzi sono legati alla “salute”, più o meno.
Il Medical Technology Enterprise Consortium (MTEC), che opera per conto del Comando di Ricerca e Sviluppo Medico dell’Esercito degli Stati Uniti, comprende tecnologie per l’editing genico, la nanotecnologia, “soluzioni di teleassistenza”, arti artificiali e impianti cerebrali. Attualmente stanno sviluppando un dispositivo indossabile per diagnosticare la Covid-19 prima della comparsa dei sintomi. Davvero. Possono etichettarti come un pericolo per la società prima che tu sia infettato da qualcosa! Governatemi meglio, per favore.
Il Medical CBRN Defense Consortium (MCDC) comprende 318 grandi e piccole imprese ed enti accademici che “sostengono i requisiti medico-farmaceutici e diagnostici del Dipartimento della Difesa (DoD) per contrastare gli agenti di minaccia chimica, biologica, radiologica e nucleare (CBRN)” e consentono “tecnologie prototipali per contromisure mediche terapeutiche mirate a bersagli virali, batterici e tossine biologiche di interesse per il DoD“, compreso lo sviluppo di vaccini.
Attraverso il meccanismo dell’Other Transactional Authority, l’MCDC ha stipulato contratti con centinaia di aziende per la fornitura di tutte le “contromisure” legate a Covid. Le iniezioni di Pfizer sono state ordinate il 20 luglio 2020 attraverso l’accordo di base tra Advanced Technologies Inc (ATI, una società di gestione dei fornitori del Dipartimento della Difesa) e Pfizer, Inc, identificato come MCDC Base Agreement No. 2020-532:
– 21 luglio 2020, MCDC Technical Direction Letter o Statement of Work (SOW) per “COVID-19 Pandemic – Large Scale Vaccine Manufacturing Demonstration” tra Pfizer e DOD/Advanced Technologies Inc.
L’esame dei contratti di Pfizer e Moderna con cui il Dipartimento della Difesa ha ordinato centinaia di milioni di iniezioni di Covid-19 ha rivelato la mancanza di una reale responsabilità per la sicurezza, l’efficacia o la qualità di produzione del prodotto da parte dei produttori farmaceutici, unita a un elevato grado di microgestione e controllo da parte dell’ente appaltante (DOD/BARDA). Sebbene si tenti di descrivere i contratti come accordi “a corta distanza“, il controllo esercitato dal Dipartimento della Difesa è schiacciante. In particolare, i contratti prevedono somme di denaro che superano qualsiasi prodotto medico legittimo esistente prodotto dalle aziende farmaceutiche.
Il contratto di Pfizer era di 2 miliardi di dollari, ma è stato esteso a circa 10 miliardi di dollari, o fino a 500 milioni di dosi. Non esiste una vera e propria responsabilità, se non lo standard di “sforzo ragionevole” applicato al produttore per la qualità o la sicurezza del prodotto. Tuttavia, gli aspetti operativi, i dati, le interazioni con la FDA e le comunicazioni del contratto sono strettamente microgestiti. Si suppone che il produttore debba tenere telefonate/riunioni quotidiane con il Dipartimento della Difesa sullo stato del progetto. Inoltre, le comunicazioni della casa farmaceutica con la FDA sono sotto stretto controllo del Dipartimento della Difesa. Non c’è la possibilità di avere un dialogo indipendente tra l’azienda farmaceutica e la FDA (cosa a cui tutte le aziende farmaceutiche sono in genere molto sensibili). Nell’ambito dei contratti OTA, tutte le comunicazioni con l’FDA sono esaminate e approvate dalla BARDA e qualsiasi incontro di persona è accompagnato da un massimo di quattro persone della BARDA.
Infine, il prodotto non è serializzato, ossia le dosi unitarie non sono codificate a barre e quindi non sono rintracciabili secondo le normali regole di distribuzione farmaceutica, che esistono per segnalare qualsiasi problema di sicurezza o qualità nella catena di fornitura. Il prodotto è quindi aperto alla falsificazione e all’adulterazione. Il prodotto viene spedito al Dipartimento della Difesa e gestito attraverso un sistema di distribuzione “black box” del Dipartimento della Difesa, apparentemente a causa dei requisiti di conservazione nella catena del freddo.
Il prodotto è considerato “proprietà del governo degli Stati Uniti” finché non viene iniettato in una persona.
Tutte le persone che svolgono mansioni lungo la produzione, la catena di approvvigionamento, la distribuzione e la somministrazione delle iniezioni sono “persone coperte” ai sensi del PREP Act e sono completamente al riparo da qualsiasi responsabilità, purché eseguano gli ordini. Indipendentemente dalla sede di lavoro, sono considerati dipendenti del governo degli Stati Uniti ai fini di questo lavoro. Inoltre, i contratti del Dipartimento della Difesa li descrivono come “applicazioni civili e militari“. Ecco la clausola PREP Act del contratto Moderna. Tutti i contratti DOD/HHS prevedono questa clausola:
È vietato eseguire test indipendenti sulle fiale per verificare la conformità del prodotto all’etichetta. Negli Stati Uniti le fiale sono “proprietà del governo americano” e i contratti d’acquisto ex-USA vietano esplicitamente il test delle fiale all’importazione.
La conclusione che emerge da questi fatti è che lo sviluppo normativo, gli studi clinici, la revisione e l’approvazione dei dati relativi ai “prototipi di contromisure” sono una messinscena, una farsa, una rappresentazione teatrale progettata per creare un falso senso di fiducia e quindi ingannare il pubblico e indurlo a iniettarsi “prototipi” che non hanno alcuna divulgazione o alcun modo per garantire la sicurezza, l’efficacia, la conformità, la purezza, la consistenza e le altre caratteristiche che ci si aspetta dai prodotti medici.
I fatti legali secondo cui qualsiasi uso di contromisure mediche coperte da EUA nell’ambito di PHE non è un’indagine clinica erano noti ai dirigenti di Pfizer che hanno firmato i contratti del luglio 2020, e anche ai funzionari del DOD/ATI e dell’HHS che hanno firmato tali contratti. Tuttavia, tutte queste parti, compresi i funzionari della FDA, hanno svolto il loro ruolo fingendo di “autorizzare” i prodotti.
Questi fatti legali riguardo le prestazioni non erano noti al pubblico – gli sperimentatori, i soggetti e l’opinione pubblica mondiale a cui veniva detto che si trattava di autentiche indagini cliniche e che i risultati dimostravano che i prodotti erano “sicuri ed efficaci“. Il pubblico degli Stati Uniti e di tutto il mondo è stato ingannato da questa finta rappresentazione teatrale.
L’opera d’arte di oggi è l’Apocalisse. Non credo che stiamo vivendo una vera e propria apocalisse, ma solo una rappresentazione teatrale messa in scena per sembrare tale. È fondamentalmente un bluff di una (piccola) cricca disperata di mostri profondamente malvagi. Non cascateci.
Fonte/tradotto da Intent to harm. L’autrice, Sasha Latypova, ha testimoniato (anche) nella sessione 124 del CIC @1:56:00(1), @3:49:50(2) espandendo sulle problematiche di produzione/qualità in relazione anche agli effetti avversi/tossicità.
David Martin: non chiamarlo vaccino, è un’arma
SARS-CoV-2: l’arma biologica controversa
COVID-19 Injections are weapons, intended to harm, Pseudo-Legalization of EUA-Covered “Military Countermeasures“ → full video here — @Doctorsforcovidethics ;
Sasha Latypova: New documents show the cover-up is even WORSE than we thought | @Redacted News Live
♦ Sasha Latypova Interview – How Pfizer & The Department Of Defense Defrauded The Public ;
♦ Un esperto di genomica scopre un contenuto interessante nelle fiale di [falso]vaccino COVID “… Questo è un elemento che aiuta a promuovere la resistenza agli antibiotici che è la spina dorsale di questi vettori … percorso di integrazione del genoma …” (@P_McCulloughMD) April 13, 2023 ;
♦ Katherine Watt and I discuss Brook Jackson v Ventavia Case Dismissal
♦ Clarification of My Message on the Global Mass Murder Campaign
♦ Cull of useless eaters. The rush to have different doses in batches was intentional actually. … @angelovalidiya ;
♦ BioNTech [e la Germania, quindi] is the Big Winner From the [fake]Vaccine Bonanza, Not Pfizer ;
↔♦ BioNTech’s 30 Billion Reasons ;
♦ Proof that the [fake]Vaccines Were a Military-Backed Countermeasure ;
♦ Covid [fake]vaccine-related deaths send ‘mortality’ to unseen highs (REPORT) ;
♦ There was intent to harm. -Dr. Rancourt ;
♦ Studi di G.Segalla, L.Frasca, … @byoblu ;
♦ The red flags in the Pfizer [fake]vaccine trial that were ignored ;
♦ Cancers are exploding. We don’t need more evidence ♦ Schroedinger’s SV40 p53 ♦ DNA contamination in the manufacturing of mRNA injectables ♦ How SV40 works … ♦ That’s a risk for genome integration, that’s a risk for cancer , that’s a risk for… ;
⚠️[passato] A livello mondiale: Inviatemi campioni anonimizzati e li sottoporrò a un test PCR e sequenzierò gli eventuali positivi per integrazione nel DNA da pseudo-vax COVID mRNA. -@P_J_Buckhaults ↔ @godlikeproductions con rif. intervista K.McKernan ↔ explosive cancers -@jcampbell
♦ You can’t inject your bioweapon and have it too! “Poison-19 injections are designed to kill, maim and reduce fertility” -@latypova ;
♦ Una libreria organizzata di oltre mille articoli sottoposti a revisione paritaria che dimostrano che i sieri magici Covid-ID19 sono dannosi. —@BorN ;
♦ The Covid “Killer Vaccine”. People Are Dying All Over the World. It’s A Criminal Undertaking [26.11.22] ;
♦ The Hard Evidence Is in: The Covid ‘Vaccine’ Is a Killer ;
♦ Denis Rancourt: interviste, presentazioni, audizioni, … ;
♦ New Study: Death Following Covid [fake]Vaccination ;
♦ Plasmid DNA Contamination: A Primer — Part 2 ;
♦ WCH to Host Follow-Up Expert Hearing on DNA Contamination in mRNA [fake]Vaccines ;
♦ Systematic Review of Autopsy Findings in Deaths after COVID-19 Vaccination ;
♦ 17+ milioni di morti causati dalle iniezioni Covid mRNA – Dr Denis Rancourt —@BorN ;
♦ Il contratto segreto UE-Pfizer sui falsi vaccini … —@GiuRos ;
♦ Embroidery: “Pfizer Vaccine -FDA Fails to … “Pfizer Evidence So Far: Coverups, … “Pfizer, FDA, CDC Hid Proven Harms … ♦ Report 93 … ;
♦ Is it possible that COVID-19 boosters trigger a cancer relapse? dr. Dalgleish ;
♦ Update – NZ Whistleblower -@igorchudov ♦ dataset on IPFS ♦ The Brook Jackson vs Pfizer case is very much alive. ♦ royalties to NIAID. ♦ Fauci admits to forcing vaccine mandates on Americans: … —@iGiuI ;
♦ Video: The Pfizer “Killer Vaccine”: “Money vs. Mortality”. Michel Chossudovsky ;
♦ 11 mila esenzioni [falsa]vaccinazione COVID/mRNA per l’élite. Ancora dubbi? ;
♦ Covid mRNA [fake]Vaccines Required No Safety Oversight – P.1 → Part.2 –@debbielerman ;
♦ Covid mRNA [fake]Vaccines Were Developed Under a Military Protocol and Required No Safety Oversight ;
♦ Why Did HHS “Partner” with DOD? -@latypova ;
♦ Why was the BioNTech/Pfizer mRNA [fake]vaccine not recalled in Feb 2021? ;
♦ Mike Yeadon: Address to parliamentary special meeting 4/12/2023 —@ICInvCo ;
♦ Expert testimony on the Pandemic in the UK Parliament ;
♦ UK COVID-19 Inquiry: Politicians Sought to Control the Scientists … ;
♦ Governments intentionally wanted to harm their people … —@C19ExpCh ;
♦ “Il 75% delle vittime della miocardite moriranno entro 10 anni.” ;
♦ RFK Jr on his new book that nails the worldwide bioweapons scandal ;
♦ Covid and Marburg Weapons Systems ;
♦ We have a pandemic of marburg and ebola lasting to at least December 31, 2028. ;
♦ There is zero contest about the DNA in the vaccines ♦ Residual DNA could be oncogenic ♦ There was no trial on the drug given to the public ♦ If you can’t address the nuclear targeting sequence in SV40 … ;
♦ Summary of Everything and Quick Links, Updated – Year End 2023 -@latypova ;
♦ Reflections on the Bret Weinstein Interview ;
♦ Gli ESENTI dal falso-vaccino anti-Covid negli USA: … [come in NZ … e in Italia chi è stato esentato?] ;
♦ Emerge una rivolta interna all’interno di Pfizer per smascherare le pratiche trascurate per approvare tutto per l’iniezione. … —@laNuovaN ;
♦ Pfizer Insider ammette che l’mRNA ucciderà miliardi di persone in pochi mesi … —@vvincvvv ;
♦ Dr.Balanzoni: Minori accompagnati….. impossibile da comprendere ma le cose stanno… ♦ … Eppure nei reparti gli anziani sono stati uccisi con dosi crescenti di midazolam … ;
♦ Self-replicating/self-amplifying RNA “vaccines” are disasters on steroids ;
↔♦ Vaccini a RNA auto-amplificante: un nuovo pericolo all’orizzonte —@MarCosent ;
↔♦ Concise explanation of the current state of the science on SARS-CoV-2, PRION, Vaccines and the warfare being waged against humanity. ;
♦“DNA found integrated in cancer cell line” – DNA integration associated with the modified mRNA shots is an issue, says Jessica Rose on Substack, and may be linked to cancer.
♦ NON sono stati commessi errori: un inno alla giustizia
♦ La FDA sapeva che le iniezioni della bioarma C19 avrebbero causato il cancro … ;
♦ Deleted Interview: Attorney Tom Renz on the Alex Jones Show ;
♦ Bill Gates Admits Testing Nanotech on Public with mRNA [fake]Vaccines ↔ @infowars –@wideawakemedia ;
♦ If Covid EUAs are legal, the virus must be a potential bioweapon ♦ Legal Context Behind the Bioweapon Hypothesis ;